


")
Our Hotline+603 - 8230 0300
Medical Device Recall
What Is Medical Device Recall?
A recall is a method of removing medical device that are in violation of laws administered by Medical Device Authority (MDA). Recall action has been set out in Section 42 of Act 737. Recall may be undertaken voluntarily and at any time by manufacturers and establishments, or at the request of the MDA.
Mandatory Recall
Mandatory recall is initiated by the authority to order for a medical device recall if a medical device possesses a high public health risk from the market as described in section 42 (4) of Act 737.
Voluntary Recall
Voluntary recall is an action taken by establishment where they are required
- to remove the medical device from the market
- to retrieve the medical device from any person to whom it has been supplied
- to notify its affected person of its defectiveness or potential defectiveness, after becoming aware that the device:
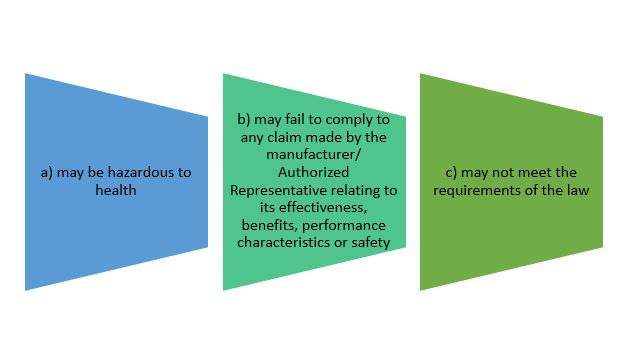
Establishment must notify authority and affected person according to recall classes before commencing recall process.
- Recall Class I, high risk, 48 hours
- Recall Class II, moderate risk, 3 days
- Recall Class III, low risk, 5 days
For further information, kindly refer to the guidance document. MDA/GD/0015 Medical Device Recall.
How To Notify Medical Device Recall?
Medical device recalls for devices that affected Malaysia can be reported via an online system, Medical Device Centralized Reporting System, MeDCReSt.
Contact Information
Vigilance Unit
MEDICAL DEVICE AUTHORITY
Ministry of Health Malaysia
Level 6, Prima 9, Prima Avenue II
Block 3547, Persiaran APEC
63000 Cyberjaya, Selangor
MALAYSIA
T: (03) 8230 0245
Updated: 20th February 2023
×



